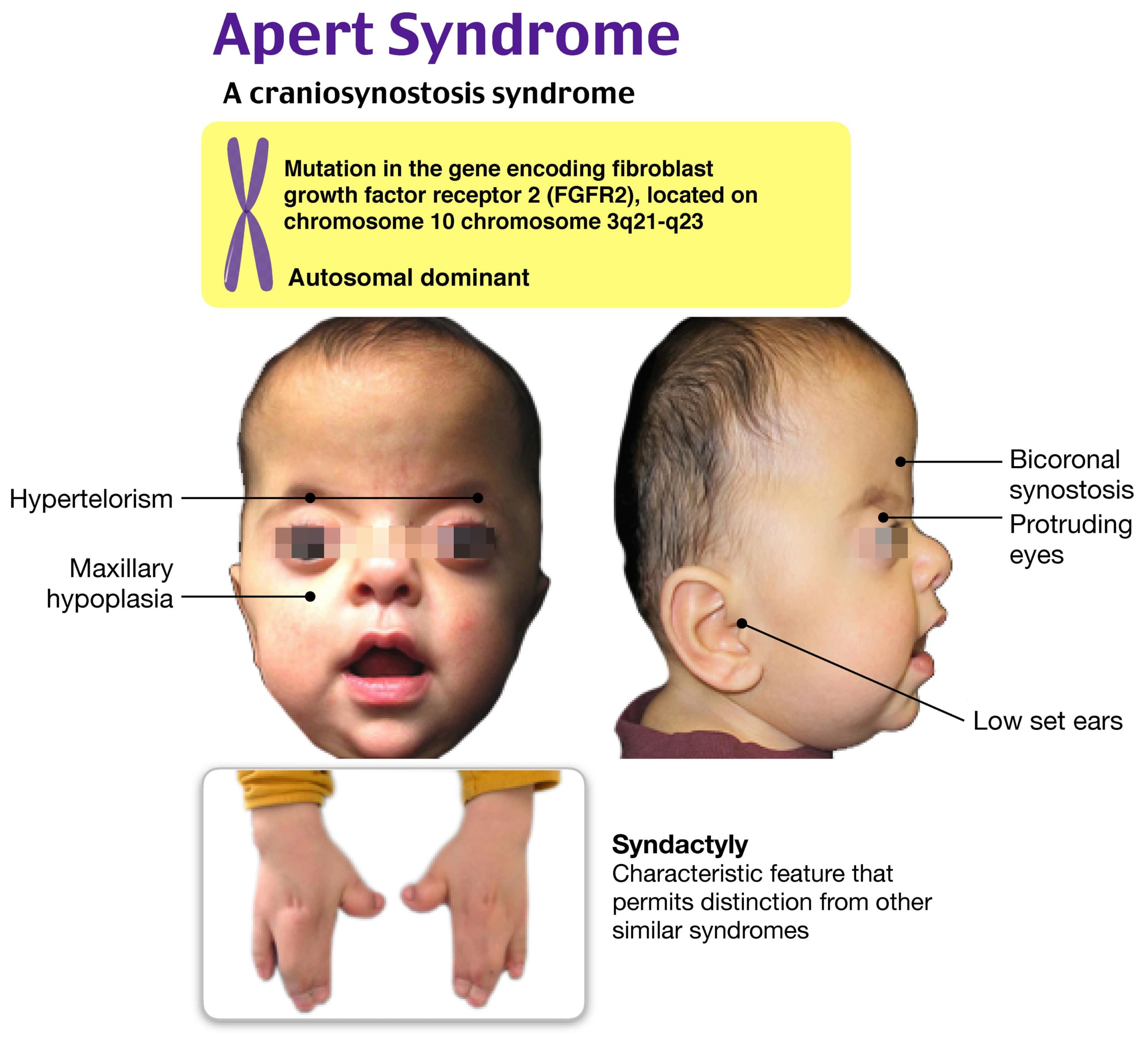
Apert syndrome is one of a class of diagnoses referred to as the acrocephalosyndactyly syndromes, which are genetic conditions characterized by multisutural craniosynostoses, midface hypoplasia with abnormal facies, and limb abnormalities, including syndactyly. The craniofacial abnormalities common among these syndromes lead to increased risk of exposure keratopathy and blindness, hydrocephalus, intracranial hemorrhage, and otolaryngologic complications. Apert syndrome is characterized by bicoronal synostosis. Facial features include hypertelorism, ptosis, and proptosis. Patients have midface hypoplasia and cleft palate and may also have choanal hypoplasia and stenosis with resultant airway obstruction. Affected children may have gaze divergence and other vision problems as well as conductive hearing deficits. Syndactyly is generally complex and symmetric, often of the middle three digits of the hands and feet. Other associated extracranial manifestations may involve the heart, kidneys, or skin. Although hydrocephalus is not common, brain anomalies and developmental or cognitive deficits are often seen.
Crouzon syndrome (B) is another cause of bicoronal synostosis, and the craniofacial findings are similar to those in Apert syndrome, including hypertelorism, proptosis, midface hypoplasia, and cleft palate. However, the extremities in patients with Crouzon syndrome are normal. Goltz syndrome (C), or focal dermal hypoplasia, is a rare X-linked dominant disorder characterized by dermal hypoplasia, aplasia cutis, nail hypoplasia, and dental anomalies. Syndactyly or ectrodactyly are associated, but craniosynostosis is not. Oculodentodigital dysplasia (D) is a type of ectodermal dysplasia that causes cardiac, neurologic, and ocular anomalies. Affected patients typically have small teeth that are susceptible to caries and a cleft lip and palate. The diagnosis is also associated with syndactyly of the fourth and fifth fingers, but not with craniosynostosis.


